Supplementary 1a: Fragmentation#
Set path to project root#
import os
import sys
def find_project_root(marker=".git"):
current_path = os.getcwd()
while current_path != os.path.dirname(current_path): # Stop at the filesystem root
if marker in os.listdir(current_path):
return current_path
current_path = os.path.dirname(current_path)
return None # Return None if the marker is not found
project_root = find_project_root()
print(project_root)
# Add project_root to the Python path
sys.path.append(project_root)
/home/yuyang/Project_local/FragBEST-Myo
Import requried packages#
import json
import MDAnalysis as mda
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import MolsToGridImage
from utils.ppseg.fragment import fragment_on_bonds
Read/write the files#
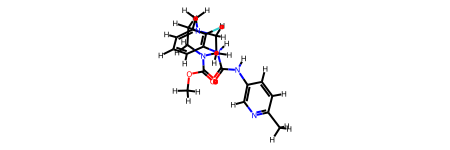
First, read the reference file (protein with ligand) using MDAnalysis. We selected the ligand by resname 2OW and convert it to rdkit.Chem.rdchem.Mol. Note that elements are needed to RDKit convert; furthermore, to make sure the connection of the atoms is correct, we used to_guess=["bonds", "elements"] in guess_TopologyAttrs().
# ------------------- start of the user-defined parameters -------------------
# set path
ref_filepath = f"{project_root}/dataset/ref/PPS_OMB_min_cg_pl.pdb"
ligand_file_path = f"{project_root}/dataset/ref/PPS_OMB_min_cg_pl_2OW_only.pdb"
# ------------------- end of the user-defined parameters ---------------------
u = mda.Universe(ref_filepath)
u.guess_TopologyAttrs(to_guess=["bonds", "elements"])
molecule = u.select_atoms("resname 2OW").convert_to("RDKIT")
/home/yuyang/Project_local/FragBEST-Myo/.pixi/envs/default/lib/python3.10/site-packages/MDAnalysis/topology/PDBParser.py:350: UserWarning: Element information is missing, elements attribute will not be populated. If needed these can be guessed using universe.guess_TopologyAttrs(context='default', to_guess=['elements']).
warnings.warn("Element information is missing, elements attribute "
Then, Use RDKit.Chem to read the molecule from a PDB file.
molecule
And, we saved the ligand only file.
u.select_atoms("resname 2OW").write(ligand_file_path)
/home/yuyang/Project_local/FragBEST-Myo/.pixi/envs/default/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1154: UserWarning: Found no information for attr: 'formalcharges' Using default value of '0'
warnings.warn("Found no information for attr: '{}'"
Fragmentation on bonds#
Next, use our developed function fragment_on_bonds to fragment the ligand by bonds. Here, we chose the bonds number 3, 7, 8, 13, 17.
# fragment the ligand molecule based on the specified bonds
mol_fragment, mol_f_idx = fragment_on_bonds(
ligand_file_path, [3, 7, 8, 13, 17], removeHs=False, with_star=False
)
Visualize the chemical fragments#
We then used RDKit’s function to visualize them in 2D space. However, the bond order is not all coverted correctly. Thus, we need to check and manually correct them when visualization.
MolsToGridImage(mol_fragment)
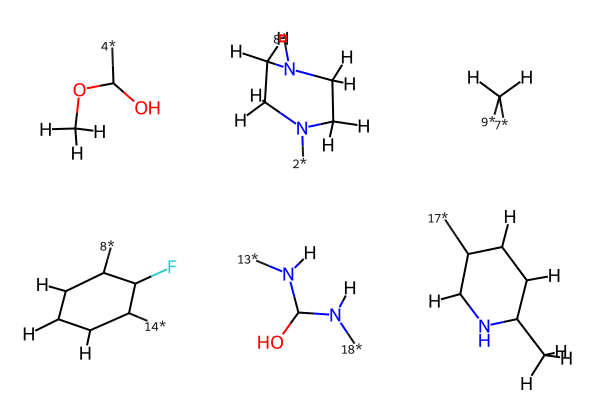
mol_fragment_smiles = [
Chem.MolToSmiles(mol).replace("(O)", "(=O)") for mol in mol_fragment
] # fixed the double bonds issues (but loss the conformation)
mol_fragment_smiles = [
each.replace("C", "c").replace("N", "n") if (idx == 3) | (idx == 5) else each
for idx, each in enumerate(mol_fragment_smiles)
] # fixed the atom type issues
mol_fragment_smiles[5] = mol_fragment_smiles[5].replace(
"(c([H])([H])[H])", "(C([H])([H])[H])"
)
mol_fragment_vis = [Chem.MolFromSmiles(smiles) for smiles in mol_fragment_smiles]
MolsToGridImage(mol_fragment_vis)
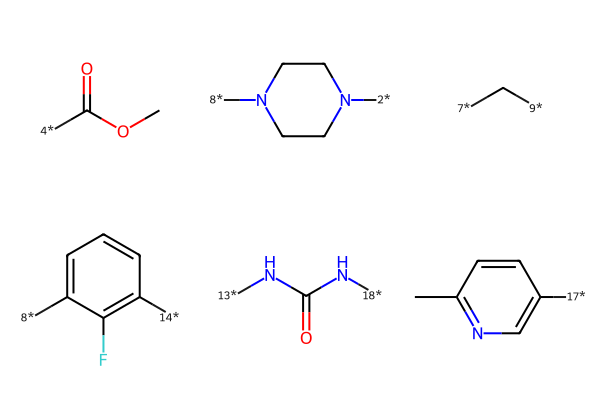
Fragment volume#
Another important information should be included is the volume of the fragments. We calculated this by RDKit.
volumes = [AllChem.ComputeMolVolume(mol) for mol in mol_fragment]
print(volumes)
[55.40000000000001, 88.88800000000002, 24.440000000000005, 85.09600000000002, 50.320000000000014, 94.30400000000002]
In our original study, each fragment volume was calculated by averaging its fragment volume in the entire ligand-bound trajectory. Thus, there is a slight difference between the provided fragment information json file (FragBEST-Myo/utils/ppseg/myo/ligand_fragments_example.json) and the one generated here.
Save fragments information#
# ----------------- start of the user-defined parameters -----------------
ligand_fragments_path = f"{project_root}/dataset/ref/ligand_fragments.json"
# ----------------- end of the user-defined parameters -------------------
labels_info = {
"0": {"name": "out of the threshold"},
"1": {
"name": "fragment 1",
"fragments_idx": mol_f_idx[0],
"fragment_vol": volumes[0],
},
"2": {
"name": "fragment 2",
"fragments_idx": mol_f_idx[1],
"fragment_vol": volumes[1],
},
"3": {
"name": "fragment 3",
"fragments_idx": mol_f_idx[3],
"fragment_vol": volumes[3],
},
"4": {
"name": "fragment 4",
"fragments_idx": mol_f_idx[4],
"fragment_vol": volumes[4],
},
"5": {
"name": "fragment 5",
"fragments_idx": mol_f_idx[5],
"fragment_vol": volumes[5],
},
"6": {
"name": "fragment 6",
"fragments_idx": mol_f_idx[2],
"fragment_vol": volumes[2],
},
}
# save the ligand fragments information
with open(ligand_fragments_path, "w") as f:
json.dump(labels_info, f, indent=2)